Biotinidase
Chemical Pathology
Notes
- Biotinidase deficiency (late-onset multiple carboxylase deficiency) is an autosomal recessively inherited disorder of biotin recycling, associated with skin rashes and neurological problems (including seizures, hypotonia, hearing loss, alopecia, developmental delay and visual problems) if left untreated.
- Biotinidase deficiency is classified as profound or partial depending on biotinidase levels. Patients with partial deficiency are largely asymptomatic unless acutely unwell.
- Biochemically, metabolic abnormalities in untreated individuals are variable but may include metabolic ketoacidosis, lactic acidosis and/or hyperammonaemia.
- Increased excretion of 3-hydroxyisovaleric, lactic and 3-hydroxypropionic acids and 3-methylcrotonylglycine may be seen on urine organic acid analysis. Acylcarnitine profiling may show marginally elevated 3-hydroxyisovalerylcarnitine.
- Early diagnosis and life-long biotin supplementation is very effective.
Sample requirements
For adults, blood taken into a 4mL EDTA tube
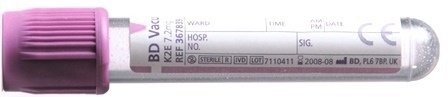
For children, blood taken into a 2mL EDTA tube
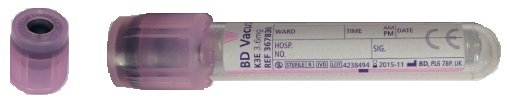
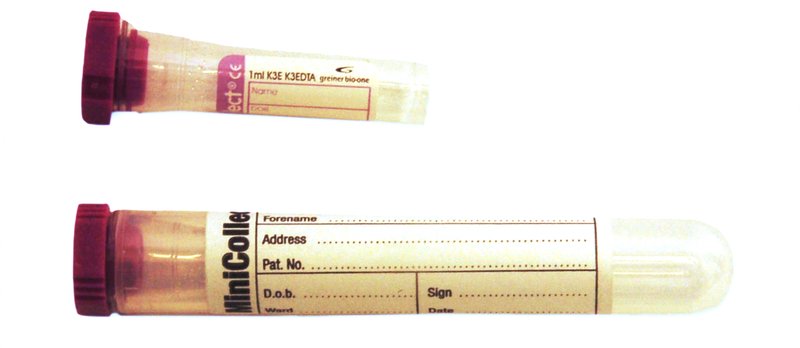
For neonates, blood taken into a 1.0mL minicollct EDTA tube
Storage/transport
Do not store. Deliver immediately to the laboratory at ambient temperature. Samples must be separated and plasma frozen within 6 hours maximum to avoid false positive results.
Required information
Please provide relevant clinical details with all requests.
Turnaround times
The samples are sent for analysis to University Hospitals Bristol NHS Foundation Trust with results expected back within 3 weeks.
Treatment should not be delayed whilst awaiting results.
Reference range
2.6 - 7.0 µmol/L/min
Further information
To learn more about biotinidase deficiency access the following document:
Technical Standards and Guidelines for the diagnosis of Biotinidase Deficiency
Page last updated: 22/11/2023